Have you ever heard of Rare Diseases? You may have but, as its name suggests, these diseases are very rare and you might not know much about them… do you? For instance, if I say “Gaucher”, “Niemann-Pick”, “Pompe”, “Fabry”… Do any of these names ring a bell with you? They are actually rare diseases or, as some researchers prefer to call them, infrequent diseases.
Rare diseases (RDs), also known as orphan diseases, are high mortality disorders with very low individual prevalence. They are the most unknown diseases for the society, and they critically affect global health due to their severity, the high number of people affected, the difficulties to find a diagnosis, and the lack of effective treatments [1,2]. Some examples of RDs are the following: Fabry, Behçet, Niemann-Pick disease, Pompe, Tay-Sachs, Sandhoff, Danon, Gaucher, Huntington, Paget and Leigh disease, Leukodystrophy, Cystic fibrosis, Mucopolysaccharidosis, Peroxisomal and Glycogen storage disorders, Prader-Willi syndrome, Wolfram, Li-Fraumeni, or the Papillon-Lefévre syndrome; among many others.
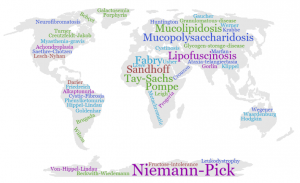
RDs are heterogeneous in nature and geographically diverse, affecting around 410 millions of people worldwide. In other words, 7% of the world population are suffering from these diseases. Around 25 million people are affected in USA, 42 million in South America, and 30 million in Europe; with 3 and 3.5 million people affected in Spain and the UK, respectively. In Europe, a disease is considered “rare” if its prevalence is less than 5 cases per 10,000 people [1,3]. Even though RDs are very infrequent, they affect more people than cancer and AIDS combined if we consider them as a whole [4].
These infrequent diseases are present throughout the whole patient’s life, being either acute, chronic, or degenerative. They can show a wide range of symptoms that tends to vary from patient to patient, which commonly leads to wrong diagnoses. RDs affect people regardless their age, although approximately 75% affect only children, who may die before they are even 5 years old. The earlier the symptoms appear, the worse the prognosis, and the higher the chances of an early death [5].
Currently, it is estimated that there are around 7,000 RDs, but this number is continuously growing. Furthermore, the increase of cases with very low prevalence is very significant, which correlates with the high variability and variety of these disorders. Although some of these RDs have their origin on the exposure to infections, toxins, or environmental causes; most of them (80%) have a genetic origin [6,7].
Today, we know that most RDs are caused by mutations in protein-coding genes involved in key cellular mechanisms, thus most of the times affecting the final protein structure, its stability, and its function. The severity of the diseases usually depends on the mutation type and the part of the gene where it occurs. This may explain why the physiopathology and symptoms of the same disease are very different from one patient to another, a very important fact to consider in this research field.

In the end, this situation mainly affects patients and families who live in constant uncertainty as they cannot be told an accurate diagnosis. This is due to the fact that the average time to get the diagnosis of one of these RDs might take around 6 to 8 years, although it can be extended to even 10 years or more. Furthermore, there is a lack of patterns that can help to identify the disease, and many physicians may have not seen this condition before. Therefore, the diagnosis can change as new or advancing symptoms appear. Nowadays, many patients, relatives, and friends who seek answers to this problem are creating and joining organizations to support other families in the same situation, which increases the visibility of these infrequent diseases [2].
The lack of effective treatments is another obstacle that people with RDs have to deal with. In 1983 in USA, due to the lack of awareness, disperse target population, and inefficient reporting regarding RDs; the Orphan Drug Act took place to encourage the development of drugs to treat them [8]. Since that date, 2,900 orphan drugs were designed, although less than 5% of RDs have a Food and Drug Administration (FDA) approved treatment. This means that at around 26.8% of patients have been receiving the wrong treatment during their lives. As a result of the reduced amount of people suffering from an individual rare disease, it is not profitable for pharmaceutical industries to invest in research focused on understanding the molecular mechanisms underlying these pathologies to develop potential drug treatments [9]. Fortunately, the number or scientific publications about RDs does not stop increasing as well as the number of researchers working on this field. , Developing new treatments for RDs, however, is still a challenge despite the continuous scientific advances, which means that more than 90% of these diseases do not have an approved treatment yet [10].
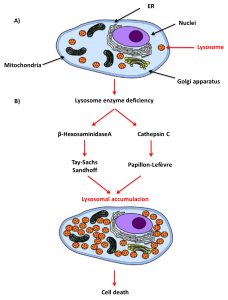
During my PhD research, I have been working with three RDs named Tay-Sachs, Sandhoff, and Papillon-Lefévre. All of them are lysosomal storage diseases (LSDs), a heterogeneous group of around 70 rare genetic diseases characterized by the accumulation of undegraded cellular products inside the lysosome due to mutations in genes encoding for lysosomal proteins (β-Hexosaminidase A and Cathepsin C, respectively)[11]. Lysosomes are acid subcellular organelles containing hydrolytic enzymes that function as the digestive and recycling system of the cell. They are involved in an essential process known as autophagy by which cellular waste is removed [12,13].
The first two, Tay-Sachs and Sandhoff are neurodegenerative diseases characterized by an early neuronal cell death because of the accumulation of GM2 gangliosides inside the lysosomes due to the deficiency in the lysosomal enzyme β-Hexosaminidase A. This enzyme is a heterodimer composed of an α-subunit and a β-subunit encoded by HEXA and HEXB gene, so mutations in these genes lead to the development of Tay-Sachs and Sandhoff diseases, respectively [14]. In the experiments I have been involved during my research, our team found a compound that improves the prognosis of the disease in vitro and in vivo. The Papillon-Lefévre is another LSD characterized by severe early-onset periodontitis and palmoplantar hyperkeratosis due to a mutation in the gene encoding for the lysosomal enzyme Cathepsin C. Part of our experimental work was involved in the design of an enzyme replacement therapy by using a recombinant enzyme produced in insect cells [15]. With these examples I have given, it is clear how these three diseases, despite having very different symptoms and being caused by mutations in different genes, have all a dysfunction in the autophagy machinery which is vital for cellular survival.
As a result of the research I have been working on with regards to LSDs, our team has proposed two approaches to improve the autophagy process and, more importantly, patients’ health. We hope that our promising results may encourage more groups to extend this research to other rare diseases.
* * *
By Beatriz Castejón Vega, postdoc at the University of Glasgow.
More information:
- Federación Española de Enfermedades Raras.
- Post “About rare diseases”, source: EURORDIS.
- Haendel, M. et al. (2019).
- Post “RARE Facts”, source: Global Genes.
- Field, M.J. & Boat, T.F. (2011).
- Nguengang Wakap, S. et al. (2019).
- Tambuyzer, E. (2010).
- Herder, M. (2017).
- Gavin, P. (2015).
- Tambuyzer, E. et al. (2019).
- Pastores, G.M. & Wang, R.Y. (2017).
- Cooper, G. M. (2000).
- Glick, D.; Barth, S.; & Macleod, K. F. (2010).
- Schuchman, E.H. & Simonaro, C.M. (2013).
- Bullón, P. et al. (2018).
- Bellettato, C. M. et al. (2018).