Déjame invitarte a un viaje al interior del cerebro. Las células más conocidas del cerebro son las neuronas. Una neurona típica tiene un cuerpo con forma de estrella con muchas prolongaciones (llamadas dendritas) que recopilan información de otras células, y una prolongación mucho más larga (llamada axón) que transporta toda esa información como impulsos eléctricos hacia otras células.
Las neuronas se han especializado tanto en la transmisión de información, que han perdido la capacidad de protegerse y obtener energía, y necesitan rodearse de células de ayuda (o auxiliares). De entre todas las células que ayudan a las neuronas a cumplir su función, los oligodendrocitos se encargan de proteger y proporcionar alimento a las neuronas. Lo hacen enrollándose alrededor de los axones de las neuronas, formando una sustancia aislante llamada mielina, de la misma forma que un cable de cobre se protege usando una vaina de plástico. Esta protección es esencial para el correcto funcionamiento de las neuronas y, si no se produce correctamente o se deteriora, puede dar lugar a enfermedades del sistema nervioso. Por ejemplo, la esclerosis múltiple se produce por la destrucción de las vainas de mielina debido a un error en el sistema inmune, que identifica a la mielina como un cuerpo extraño (como si fuera un agente infeccioso) y lo ataca. Esta pérdida de mielina puede llevar a una desprotección prolongada de los axones de las neuronas, lo cual hace que funcionen mal y, finalmente, que mueran por falta de nutrientes.
Otro ejemplo es la investigación que desarrollé durante mi doctorado. Ésta se enfocó en cómo los oligodendrocitos podrían ser una parte fundamental en el desarrollo de la enfermedad llamada esclerosis lateral amiotrófica (también conocida como ELA). Sí, es un nombre bastante largo, pero déjame que te explique en qué consiste esta devastadora enfermedad: Hay unas neuronas especializadas en activar la contracción de nuestros músculos para que nos podamos mover. Se las conoce como neuronas motoras. Estas neuronas son muy largas: Van desde la médula espinal, que va por dentro de las vértebras de la espalda, hasta los músculos que activan. Por ejemplo, las que activan los músculos de tus brazos tienen axones que van desde debajo de tu cuello hasta el brazo, y las que activan los músculos del pie pueden ir desde la parte más baja de tu espalda hasta la punta del pie. ¡Una sola célula puede medir casi un metro!
Las neuronas motoras de la médula espinal normalmente necesitan ser activadas por otras neuronas que están en el cerebro, en una zona llamada corteza motora. Si algo hiciera que algunas de estas neuronas tuviesen problemas para activar a las neuronas de la médula espinal, no podrías mover los músculos que éstas activasen. Esto es precisamente lo que se observa en pacientes con ELA: Normalmente comienzan no pudiendo mover correctamente brazos o piernas, pero a medida que la enfermedad se desarrolla, también degeneran las neuronas motoras que activan los músculos que permiten el habla o tragar (como la lengua) y, en estado terminal, respirar (al afectarse los músculos del pecho y el diafragma). Todo este proceso suele llevar alrededor de 3 a 5 años y es devastador y muy doloroso tanto para el paciente como para la gente que le rodea. Los investigadores estamos extremadamente agradecidos a los muchos pacientes con ELA que han donado su cerebro para permitirnos investigar cómo se produce la enfermedad. Gracias a esos cerebros, pudimos aprender que las neuronas motoras de estos pacientes van muriendo y desaparecen a medida que la enfermedad avanza. Una pregunta que tuvo a muchos investigadores intrigados era por qué, de entre todos los tipos de neuronas, son las neuronas motoras las que mueren principalmente en pacientes con ELA. ¿Qué tienen de especial esas neuronas que las haga más vulnerables que el resto? Lo dijimos antes: Su longitud.
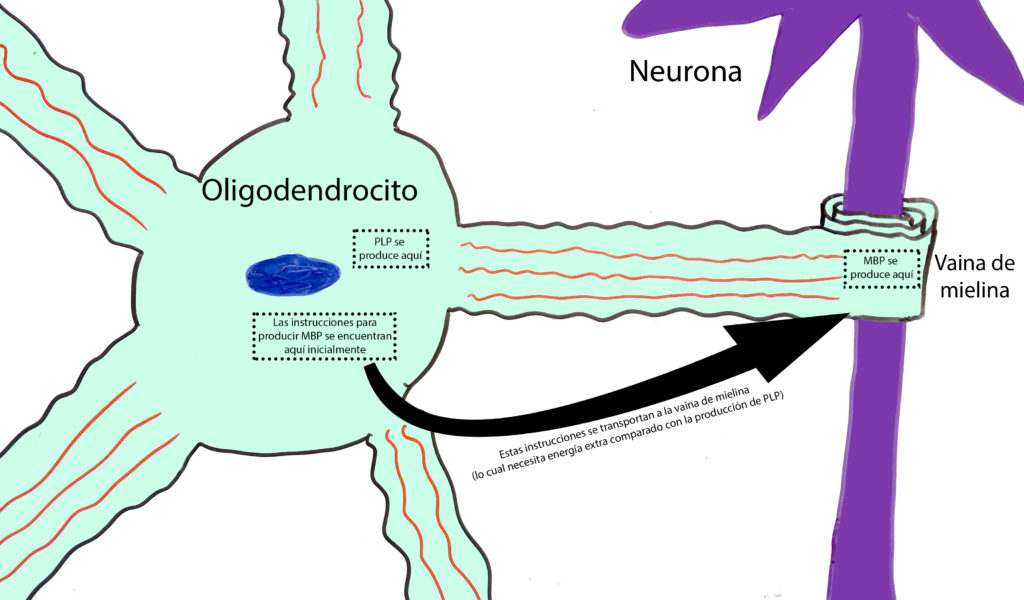
Estas células tan largas requieren muchísimas células de ayuda a lo largo de su axón para obtener el aislamiento y los nutrientes necesarios de los cuales obtener energía para funcionar. Como os podéis imaginar, mantener a tantas células de soporte activas requiere de una gran cantidad de energía. Mi investigación doctoral en el Sheffield Institute for Translational Neuroscience (SITraN) se centraba en averiguar si los procesos celulares de los oligodendrocitos que requieren una mayor cantidad de energía funcionan con la misma eficiencia que los procesos que no requieren de tanta energía en el transcurso de la ELA. Recordemos que los oligodendrocitos envuelven las prolongaciones de las neuronas con mielina. Esta mielina está formada principalmente de grasa, pero también de un buen número de proteínas, principalmente dos: La proteína proteolípida (PLP, por sus siglas en inglés) y la proteína básica de la mielina (MBP, por sus siglas en inglés).
Los oligodendrocitos tienen muchas prolongaciones (como brazos), y la mielina se encuentra al final de cada prolongación. La PLP se produce en el centro de la célula y luego se transporta a la prolongación. Sin embargo, la MBP se produce en la prolongación. Esto quiere decir que las instrucciones para su síntesis deben transportarse a lo largo de todo el ͞brazo͟ del oligodendrocito, requiriendo de una mayor cantidad de energía. Por lo tanto, si estas células no estuvieran provistas de la suficiente cantidad de energía, no se podría sintetizar tanta MBP comparado con la cantidad de PLP, la cual no requiere tanta energía para su síntesis. Lo que hemos descubierto es que, en efecto, la cantidad de PLP alrededor de las neuronas motoras es la misma en cerebros con ELA que en cerebros sanos, pero no así la cantidad de MBP, la cual se ve muy reducida en cerebros con ELA. Esto le da fuerza a nuestra hipótesis de que los oligodendrocitos no pueden obtener suficiente energía en los cerebros de pacientes con ELA. Sin embargo, harían falta más pruebas para comprobar que este efecto no ocurre por alguna otra razón como, por ejemplo, que la MBP se degrade después de haberse producido.
Al fin y al cabo, los cerebros que examinamos, aunque proporcionan información muy útil, no dejan de ser una imagen de cómo es la enfermedad en su momento final, lo cual no nos permite en la mayoría de los casos ver cómo se desarrolla la enfermedad a nivel celular a lo largo de la vida del paciente. A los investigadores nos es imposible obtener muestras cerebrales en diferentes estadios de la enfermedad, ya que esto dañaría el cerebro del paciente. Para poder investigar la enfermedad en el tiempo, hacen falta modelos animales (como ratones o pez cebra) que desarrollen la enfermedad para después poder hacerles un seguimiento continuo desde el nacimiento. Es cierto que aun queda mucha investigación que hacer para resolver todos los enigmas que esta enfermedad presenta y que la ciencia avanza siempre a pequeños pasos. Sin embargo, también es cierto que si hoy en día tenemos un mejor entendimiento de cómo funcionan la ELA y otras enfermedades neurodegenerativas, es gracias a esa gran cantidad de pequeños pasos dados previamente por miles de investigadores y pacientes. Gracias a ellos, cada día estamos un poco más cerca de hallar una forma de frenar o incluso curar estas enfermedades.
Por Dr Alejandro Lorente Pons . Sheffield Institute for Translational Neuroscience, University of Sheffield, Sheffield. Teaching associate, School of Biochemistry, University of Bristol, Bristol. Delegación de CERU Southwest.