- Los grandes conjuntos de datos genómicos son clave para comprender la evolución de las especies, aunque su análisis puede ser lento y requiere mucho poder computacional.
- Un equipo dirigido por investigadores de la Universidad Queen Mary de Londres, la Universidad de Bristol y la University College London ha desarrollado un método más eficiente para estimar cronologías evolutivas utilizando conjuntos de datos genómicos.
- Usando su novedoso método computacional, los investigadores han confirmado que los grupos de mamíferos placentarios se originaron después de la extinción masiva del Cretácico-Paleógeno, dando así una nueva perspectiva a un tema que lleva mucho tiempo debatiéndose en biología evolutiva.
La investigación describe un nuevo y rápido método computacional que puede usarse para obtener con una mayor precisión árboles filogenéticos con información de tiempo absoluto, también conocidos como “árboles calibrados”. Los autores han utilizado este novedoso método para analizar un conjunto de datos genómicos de grupos de mamíferos placentarios modernos con el objetivo de determinar si su origen tuvo lugar antes o después de la extinción masiva del Cretácico-Paleógeno (K-Pg), la cual acabó con más del 70% de todas las especies, incluidos todos los dinosaurios.
Los hallazgos confirman que los antepasados de los grupos de mamíferos placentarios modernos son posteriores a la extinción del K-Pg que ocurrió hace 66 millones de años, resolviendo así la gran polémica sobre los orígenes de los mamíferos modernos. En concreto, los mamíferos placentarios, entre los cuales se incluyen los humanos, son el grupo más diverso de mamíferos existentes hoy en día e incluyen grupos morfológicamente muy distintos tales como primates, roedores, cetáceos, carnívoros, quirópteros (murciélagos); entre muchos otros.
El equipo de investigación fue dirigido por el Dr. Mario dos Reis (Queen Mary University of London) y el Profesor Phil Donoghue (University of Bristol), e incluyó científicos de Queen Mary, University of Bristol, University College London (UCL), Imperial College London y la University of Cambridge.
La Dra. Sandra Álvarez-Carretero, autora principal del artículo de UCL (parte de Queen Mary cuando comenzó la investigación de este estudio), dice: “Tras integrar genomas completos y la información fósil necesaria, pudimos reducir la incertidumbre de este análisis y obtener con precisión el árbol calibrado de mamíferos. ¿Coexistieron los grupos de mamíferos placentarios modernos con los dinosaurios o se originaron después de la extinción masiva? ¡Ahora tenemos una respuesta definitiva! «
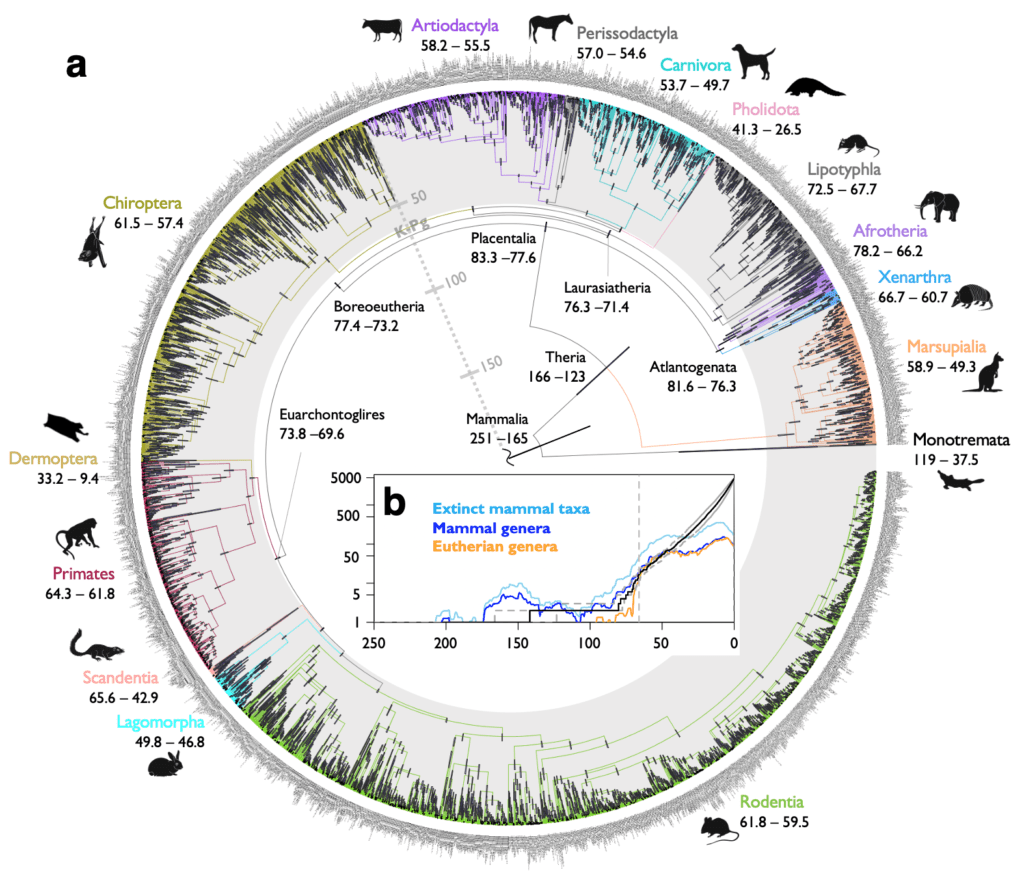
“La cronología de la evolución de los mamíferos es quizás uno de los temas más polémicos de la biología evolutiva. Los primeros estudios estimaron que el origen de grupos placentarios modernos tuvo lugar durante el Cretácico temprano, durante la era de los dinosaurios. Los estudios publicados durante las últimas dos décadas han mantenido un constante debate alrededor de dos escenarios de diversificación: antes o después del K-Pg. Nuestra cronología estimada con precisión resuelve este problema.», añade el profesor Donoghue, coautor principal del artículo.
Dado que los proyectos de secuenciación que se están llevando a cabo en todo el mundo están produciendo cientos o miles de secuencias genómicas, y habiendo planes inminentes para secuenciar más de un millón de especies, los biólogos evolutivos pronto tendrán una gran cantidad de información en sus manos. Sin embargo, los métodos actuales para analizar los vastos conjuntos de datos genómicos disponibles y crear líneas de tiempo evolutivas son ineficientes y computacionalmente costosos.
“Inferir árboles calibrados es un objetivo fundamental en biología. Sin embargo, los métodos más avanzados requieren el uso de ordenadores para simular líneas de tiempo evolutivas y evaluar las más plausibles. En nuestro caso, esto fue difícil debido al gigantesco conjunto de datos analizados, el cual involucra datos genéticos de casi 5.000 especies de mamíferos y 72 genomas completos.”, dice el Dr. dos Reis.
En este estudio, los investigadores desarrollaron un nuevo y rápido método Bayesiano para analizar un gran número de secuencias genómicas, el cual también integra las incertidumbres que estos datos puedan albergar. “Resolvimos los obstáculos computacionales dividiendo el análisis en subpasos: primero, simulando líneas de tiempo usando los 72 genomas y, después, usando los resultados correspondientes para guiar las simulaciones usando las especies restantes. El uso de genomas reduce la incertidumbre porque la información que contienen permite que las líneas de tiempo no plausibles que se obtienen en las simulaciones sean rechazadas.» dice el Dr. dos Reis.
“Nuestra línea de procesamiento de datos, lo que denominamos pipeline, obtuvo datos genómicos de varias bases de datos para tantas especies de mamíferos como fuera posible. Esto fue un desafío porque las bases de datos genéticas no son perfectas y contienen inexactitudes, motivo por el cual tuvimos que desarrollar una estrategia para identificar muestras de mala calidad o datos mal etiquetados, los cuales teníamos que eliminar.”, agrega el Dr. Asif Tamuri, coautor principal del artículo de UCL, responsable de ensamblar el conjunto de datos genómicos de mamíferos.
Usando su novedoso método, el equipo de investigadores pudo reducir el tiempo de computación para este complejo análisis de décadas a meses. “Si hubiéramos intentado analizar este gran conjunto de datos de mamíferos en una supercomputadora sin utilizar el método Ba ayesiano que hemos desarrollado, habríamos tenido que esperar décadas para inferir el árbol calibrado de mamíferos. ¡Imaginad cuánto tiempo podría llevar este análisis si usáramos nuestros propios ordenadores!”, dice la Dra. Álvarez-Carretero. “Además, también logramos reducir el tiempo de cómputo en un factor de 100. Este nuevo método no solo permite el análisis de conjuntos de datos genómicos, sino que, al ser más eficiente, también reduce sustancialmente las emisiones de CO2 liberadas debido a la computación.”, la Dra. Álvarez-Carretero continúa.
El método desarrollado en el estudio podría usarse para resolver otras líneas de tiempo evolutivas problemáticas que requieran el análisis de grandes conjuntos de datos genómicos. Usando este nuevo método Bayesiano con los genomas que van a estar disponibles una vez sean secuenciados como parte de los proyectos Darwin Tree of Life y Earth BioGenome, la idea de estimar una escala de tiempo evolutiva con mayor precisión y menos incertidumbre para el Árbol de la Vida parece ahora al alcance.
La investigación fue financiada por el Consejo de Investigación de Biotecnología y Biociencias (BBSRC, siglas en inglés) de Reino Unido.
Puedes lee este artículo a través de este enlace.
El texto ha sido traducido de la versión original en inglés por Laura Martínez Maestro y Sandra Álvarez-Carretero.


